
Quotation: Hoang N, Sardaripour N, Ramey GD, Schilling Ok, Liao E, Chen Y, et al. (2024) Integration of estimated regional gene expression with neuroimaging and medical phenotypes at biobank scale. PLoS Biol 22(9):
e3002782.
https://doi.org/10.1371/journal.pbio.3002782
Tutorial Editor: Alex Fornito, The College of Melbourne, AUSTRALIA
Obtained: March 22, 2024; Accepted: August 1, 2024; Revealed: September 13, 2024
Copyright: © 2024 Hoang et al. That is an open entry article distributed beneath the phrases of the Creative Commons Attribution License, which allows unrestricted use, distribution, and replica in any medium, supplied the unique creator and supply are credited.
Information Availability: All related information are included within the Supporting Information information and as a part of our browser-based software: https://github.com/nhunghoang/twas-webapp
Funding: This work was supported by the Nationwide Institutes of Well being (1RF1MH125933 to MR; R35GM127087 to JAC; R01HG011138 to ERG; R01GM140287 to ERG) and the Nationwide Science Basis (2207891 to MR). The funders had no position in research design, information assortment and evaluation, choice to publish, or preparation of the manuscript.
Competing pursuits: The authors have declared that no competing pursuits exist.
Abbreviations:
DLPFC,
dorsolateral prefrontal cortex; FDR,
false discovery charge; GTEx,
Genotype-Tissue Expression Challenge; GWAS,
genome-wide affiliation research; HCP,
Human Connectome Challenge; SNP,
single-nucleotide polymorphism; TWAS,
transcriptome-wide affiliation research
Introduction
A lot of human neuroscience seeks to grasp the organic foundation of particular person variation in mind group [1–6]. Research have proven that this variation is steady over time [7,8], predicts operate or habits [9,10], and may act as a fingerprint of wholesome [11,12] and diseased [13,14] mind states. They’ve additionally proven that a lot of this variation is strongly heritable and due to this fact genetically encoded [15–18]. Individually, complementary research have proven the presence of correlated variation in gene expression and neural group throughout mind areas [19–27]. Collectively, this literature motivates the necessity for integrative analyses of mind individuality throughout individuals and mind areas.
Such integrative analyses finally require information on genomes, brain-wide gene expression, in addition to neuroimaging and medical phenotypes in the identical human populations. Correspondingly, such analyses are hampered, at current, by the dearth of those multifaceted information. As an alternative, the genetic foundation of particular person variation in neuroimaging phenotypes is primarily investigated with genome-wide affiliation research (GWAS) [16–18,28–32]. Distinguished examples of those research have used information from the ENIGMA Consortium [33,34], the UK Biobank [35–37], and the ABCD Challenge [38]. These research have linked variation in phenotypes to single-nucleotide polymorphisms (SNPs), variants of DNA base pairs at particular positions within the genome. Strengths of those research embody the power to scan complete genomes and to straight uncover nucleotide-level underpinnings of neuroimaging phenotypes. Limitations of those research embody the lack to disambiguate correlated affiliation patterns of adjoining SNPs (recognized in genetics as linkage disequilibrium) and, extra usually, to determine organic mechanisms of variation in neuroimaging phenotypes. In addition they embody the necessity to take a look at tens of millions of associations (1 take a look at for every pair of SNP and phenotype) and the ensuing burden on statistical energy necessitated by stringent correction for these many checks. In apply, sturdy GWAS for a lot of advanced phenotypes, reminiscent of top or blood stress, can require samples from tens of millions of individuals [39–41]. The prices of imaging the mind, nevertheless, make it unimaginable to accumulate samples of this measurement in neuroimaging analysis [42]. Collectively, these limitations have left gaps in present analyses of human mind individuality.
Right here, we assist to bridge these gaps by estimating genetically regulated gene expression, or gr-expression, throughout cortical and subcortical mind areas. Gene expression is regulated by a number of genetic and environmental elements. Our estimation focuses on considered one of these elements, genetically encoded components which might be near the gene alongside the linear genome (cis-genetic regulation) [43]. We don’t take into account different elements, together with genetically encoded components removed from the gene (trans-genetic regulation), in addition to environmental elements. The genetics literature contains quite a lot of strategies for estimating regional gr-expression from genetic information [44,45]. Our research makes use of Joint-Tissue Imputation, a state-of-the-art methodology that trains linear regression fashions of gr-expression on straight measured gene expression from postmortem samples [43].
We used this estimated gr-expression to carry out transcriptome-wide affiliation research or TWAS. We particularly related Joint-Tissue estimates of gr-expression with neuroimaging phenotypes and brain-related medical phenotypes. TWAS observe the identical methodology as GWAS, besides that they hyperlink variation of neuroimaging phenotypes to regionally particular gr-expression of genes, quite than to regionally agnostic variation of SNPs. TWAS have a number of benefits over GWAS: they combine indicators throughout a number of SNPs, present interpretable outcomes on the degree of genes, are much less inclined to linkage disequilibrium, and require many fewer statistical checks. Nevertheless, TWAS are additionally restricted to genes with obtainable estimates of regional gr-expression and, like GWAS, are finally affiliation research that can’t alone set up causal results of genes on phenotypes.
TWAS are widespread within the wider genomics literature [44–48] however, regardless of their benefits, are uncommon in neuroimaging genomics. We hypothesize that one main motive for his or her lack of adoption lies within the comparatively theoretical nature of their enchantment to neuroimaging researchers. First, the oblique nature of estimated gr-expression could make it tough to narrate this amount to straight assayed gene expression of regional transcriptomic research. Second, the equally oblique nature of TWAS could make it tough to determine the sensible benefits of those research relative to the extra established GWAS. For instance, the few present TWAS of neuroimaging phenotypes within the literature [49–52] haven’t benchmarked these analyses in opposition to GWAS. Third, and associated to those limitations, the sector lacks built-in sources that hyperlink associations of regional estimates of gr-expression and SNPs on the one hand, to neuroimaging and medical phenotypes alternatively.
We suggest that overcoming these limitations might help facilitate the adoption of TWAS in neuroimaging genomics. Right here, we assist to take action through the use of estimated gr-expression to combine large-scale genomic, transcriptomic, neuroimaging, and medical information units. First, we confirmed that patterns of estimated gr-expression recapitulate mind regional identities and inter-regional correlation construction of straight assayed gene expression. Second, we used these estimates to carry out TWAS of gr-expression and gray-matter volumes within the UK Biobank information set [35–37]. We straight benchmarked these TWAS in opposition to GWAS to point out broad similarities but additionally vital variations within the interpretability and statistical energy of those approaches. Third, we built-in our outcomes with an impartial TWAS of brain-related medical phenotypes from BioVU, the Vanderbilt Biobank [53]. This integration linked SNPs and genes to neuroimaging and medical phenotypes by way of associations with estimated gr-expression. Fourth, we constructed polygenic fashions of gr-expression to find associations of gr-expression with neuroimaging phenotypes within the Human Connectome Challenge (HCP) [54], a small neuroimaging-genomic information set with high-quality practical imaging information. Lastly, we confirmed that estimates of gr-expression had been replicable in an impartial information set. We additionally confirmed that magnitudes of TWAS had been usually replicable whereas p-values of TWAS had been replicable in giant samples of the UK Biobank. We developed a browser-based software for interactive exploration of our multifaceted affiliation outcomes. Collectively, our analyses assist to facilitate the adoption of TWAS in neuroimaging genomics.
Outcomes
Estimation of genetically regulated gene expression throughout mind areas at biobank scale
We used Joint-Tissue Imputation [43], a just lately developed state-of-the-art methodology from computational genomics, to estimate the genetically regulated expression of 18,647 genes throughout 10 cortical and subcortical mind areas for 45,549 individuals from the UK Biobank (64 ± 7.7 years outdated, 52% feminine) and 657 individuals within the HCP (29 ± 3.6 years outdated, 52% feminine).
Joint-Tissue Imputation fashions estimate genetically regulated gene expression (gr-expression) as a weighted linear mixture of SNPs which might be near the gene of curiosity alongside the linear genome. These fashions study weights for every tissue–gene pair by coaching on genetic sequences and straight measured gene expression from postmortem samples (Fig 1A). Joint-Tissue Imputation leverages shared patterns of genetic regulation throughout mind areas to enhance the estimation of gr-expression in particular person areas. On this means, this methodology extends and generalizes PrediXcan, a pioneering estimation methodology that fashions gr-expression by coaching fashions solely on expression information from the mind area of curiosity [55].
Fig 1. Estimation of genetically regulated gene expression from genetic information.
(A) Pipeline for estimation of gr-expression with Joint-Tissue Imputation. Left: Joint-Tissue Imputation fashions are skilled on genetic sequences and straight assayed gene expression from postmortem mind samples within the GTEx and PsychEncode initiatives. Heart: The fashions are skilled to estimate gr-expression as a weighted sum of SNPs which might be near the gene of curiosity alongside the linear genome. The estimation contains elastic-net regularization as a result of the variety of these SNPs sometimes exceeds the variety of samples within the coaching information. Proper: The skilled fashions had been used to estimate gr-expression from genetic sequences of neuroimaging-genomic samples within the UK Biobank and the HCP. (B) An illustration of the ten cortical and subcortical areas with obtainable fashions of gr-expression. Numbers in parentheses check with all fashions that handed baseline efficiency thresholds for the prediction of noticed gene expression on held-out information (r2 > 0.01 and pFDR < 0.05). (C, D) Predictive efficiency of gr-expression fashions on held-out information from the GTEx information set. (C) Histograms of r [2], the variance of straight assayed gene expression defined by estimated gr-expression. (D) Histograms of p-values (−log10 pFDR) on these r2 values. Areas are coloured as in panel B. FDR, false discovery charge; GTEx, Genotype-Tissue Expression Challenge; HCP, Human Connectome Challenge; SNP, single-nucleotide polymorphism.
In our research, we used Joint-Tissue Imputation fashions that had been beforehand skilled on whole-genome sequences and gene-expression information from 838 mind samples within the Genotype-Tissue Expression Challenge (GTEx) [56]. The samples comprise 10 cortical and subcortical areas (Fig 1B). To check the replicability of our analyses, we moreover used the identical fashions skilled on sequencing and expression information from 415 impartial samples of the dorsolateral prefrontal cortex (DLPFC) within the PsychENCODE Challenge [57]. Collectively, we thought of 94,345 Joint-Tissue Imputation fashions, or all performant brain-regional fashions at the moment obtainable within the literature.
Joint-Tissue Imputation fashions have been extensively validated in earlier work [44–48]. This validation included quantifying the connection of gr-expression to straight assayed expression. On this research, we adopted all fashions of gr-expression that handed baseline efficiency thresholds for the prediction of noticed gene expression on held-out information (r2 > 0.01 and pFDR < 0.05). In apply, the predictive efficiency of gr-expression fashions spanned a variety (Fig 1C and 1D). Low predictive efficiency doesn’t essentially imply that the fashions are inaccurate as a result of the genetic regulation of gene expression—the higher certain on predictive efficiency—varies significantly for particular person genes. Furthermore, comparatively low associations between gr-expression and assayed expression are greater than offset by good points in statistical energy of transcriptome-wide affiliation analyses, as we describe beneath.
Genetically regulated gene expression recapitulates the group of straight assayed gene expression
We started by testing the extent to which gr-expression recapitulated present information of genetic-ancestry relationships, brain-regional identities, in addition to inter-regional correlations of straight assayed gene expression.
First, we examined if gr-expression patterns mirrored recognized genetic-ancestry relationships from the ethnically numerous pattern of the UK-Biobank cohort (Strategies, S1 Table). Genetic ancestry denotes genetic commonalities inside teams of individuals however doesn’t essentially replicate genealogical ancestry (household strains) or self-reported ethnicity. We adopted commonplace apply to estimate genetic ancestry utilizing principal element evaluation of gene information. We particularly used principal element evaluation to generate low-dimensional embeddings of brain-wide gr-expression from every individual (utilizing the individuals × [brain-wide gr-expression] matrix). As anticipated, this evaluation partitioned individuals into clusters of African, Asian, and European populations with gradients between these clusters reflecting recognized patterns of genetic admixture (Fig 2A). This embedding displays patterns of genetic ancestry which might be recognized and had been beforehand described in analyses of genetic-sequence information [58].
Fig 2. Genetic ancestry, regional identification, and inter-regional group of estimated gr-expression.
(A, B) Principal element embeddings of estimated gr-expression from the ethnically numerous pattern of the UK-Biobank cohort (S1 Table). (A) An embedding of brain-wide gr-expression: scatter plots of principal elements of the individuals × [brain-wide gr-expression] matrix, the place individuals denote individuals from the UK-Biobank pattern and brain-wide gr-expression denotes brain-wide estimates of gr-expression for all genes that had Joint-Tissue Imputation fashions for every of the ten areas. (B) An embedding of regional gr-expression: scatter plots of principal elements of the areas × [regional gr-expression] matrix the place areas denote the ten areas of individuals from the UK-Biobank pattern and regional gr-expression denotes regional estimates of gr-expression for all genes that had Joint-Tissue Imputation fashions for every of those areas. (C–Ok) A 3 × 3 matrix of plots of inter-regional coexpression: correlations between straight assayed expression and estimated gr-expression. The primary row and column present outcomes on straight assayed gene expression information from the Allen Human Mind Atlas. The second row and column present outcomes on straight assayed gene expression information from the GTEx challenge. The third row and column present outcomes on estimated gr-expression from the ethnically numerous pattern of the UK-Biobank pattern. (C, G, Ok) Associations between inter-regional coexpression and Euclidean distance in every information set. (D, E, H) Associations between inter-regional coexpression throughout information units. P-values denote the chance of acquiring coexpression of a minimum of equal magnitude in information with preserved correlation coefficients between coexpression and Euclidean distance (estimated from 10,000 random samples). (F, I, J) Heatmaps of inter-regional coexpression, averaged throughout individuals in every information set (regional numbers observe numbers in panel B). DLPFC, dorsolateral prefrontal cortex; GTEx, Genotype-Tissue Expression Challenge.
Second, we examined if gr-expression patterns mirrored regional mind identities throughout individuals in the identical pattern. For this evaluation, we generated principal element embeddings of particular person region-specific gr-expression (utilizing the areas × [regional gr-expression] matrix). This evaluation partitioned gr-expression into well-delineated regional clusters and revealed anatomically interpretable teams of cortical, limbic, and basal ganglionic clusters (Fig 2B). Collectively, these outcomes present that gr-expression concurrently displays genetic-ancestry identities throughout individuals and brain-regional identities inside individuals. They suggest, particularly, that associations of gr-expression, or TWAS, can seize variation throughout individuals, equally to GWAS, in addition to variation throughout areas, equally to regional transcriptomic research.
Third, we in contrast inter-regional correlations of estimated gr-expression to inter-regional correlations of straight assayed expression information from the Allen Human Mind Atlas and the GTEx Challenge. Latest research have proven that inter-regional coexpression exponentially decays as a operate of inter-regional distance [59,60]. We reproduced these relationships by displaying sturdy inverse nonlinear relationships between inter-regional coexpression within the Allen and GTEx information and Euclidean distance: Allen versus distance rspearman = −0.711 and GTEx versus distance rspearman = −0.721 (Fig 2C and 2G). We discovered the same, albeit weaker, relationship within the estimated gr-coexpression information: UK Biobank versus distance rspearman = −0.480 (Fig 2K). Extra straight, we discovered sturdy linear relationships between the inter-regional coexpression within the Allen and GTEx information: Allen versus GTEx rpearson = 0.683 (Fig 2D). We discovered comparable relationships between estimated and straight assayed inter-regional coexpression: UKB versus Allen rpearson = 0.613 and UKB versus GTEx rpearson = 0.861 (Fig 2E and 2H). Heatmaps of all coexpression patterns mirrored associations between cortical, basal ganglionic, and different subcortical methods (Fig 2F, 2I and 2J). Lastly, we confirmed that the connection of coexpression with distance was not ample to elucidate these similarities of coexpression (p ≤ 0.005 for all checks).
Collectively, these outcomes present multifaceted assist for the organic validity, anatomical interpretability, and sensible utility of estimated gr-expression. On this means, they set up a basis for using gr-expression in neuroimaging TWAS.
TWAS hyperlink genetically regulated gene expression with regional gray-matter volumes
We hypothesized that the combination of a number of SNPs into fashions of regional gr-expression would enable us to detect novel and neurobiologically significant associations. To check this speculation, we carried out TWAS to determine associations between particular person variation of regional gr-expression and gray-matter volumes (Fig 3A). Grey-matter volumes are heritable phenotypes which were linked to many genetic variants in earlier GWAS [16,17,31]. We centered our affiliation research on 8 areas with obtainable FreeSurfer [61] segmentations and due to this fact excluded substantia nigra and hypothalamus from subsequent analyses (see Strategies for regional definitions).
(A) A pipeline for transcriptome-wide affiliation research, or TWAS, of neuroimaging phenotypes. The inputs to TWAS comprise values of regional gr-expression (left) and regional phenotypes (proper), estimated in the identical individuals. The outputs are associations between the person variation of regionally particular gr-expression and neuroimaging phenotypes throughout individuals (heart). (B) Inside-regional associations of gr-expression and gray-matter volumes for two consultant areas. Every level denotes an affiliation between the person variation of gr-expression and quantity in the identical area. The horizontal axis exhibits the chromosome location of particular person genes. The vertical axis exhibits the p-values (–log10 p) of associations. Stable-color factors signify associations that go the thresholds of pFDR = 0.05 or pBonferroni = 0.05 (horizontal strains). Supply information could be present in S2 Table. (C) Associations between SNP-based GWAS and gene-based TWAS for two consultant areas. Left: Scatter plots of p-values (–log10 p) for associations of all genes and SNPs. These plots protect all genes and SNPs however lack the one-to-one relationship between genes and SNPs. Proper: Corresponding scatter plots for the best-performing genes and SNPs. Every gene in TWAS matches with its best-performing SNP in GWAS. Equally, every SNP in GWAS matches with its best-performing gene in TWAS. These plots present one-to-one relationships however exclude many genes and SNPs. (D) Numbers of associations (pFDR < 0.05 or pBonferroni < 0.05) detected with TWAS and GWAS. Stable colours denote numbers of associations detected with TWAS alone. Beige colours denote variety of genes detected with GWAS alone. Stripe patterns denote numbers of genes detected with each TWAS and GWAS. The highest bar for every area adopts an FDR correction for TWAS associations (pFDR < 0.05), whereas the underside bar adopts a stricter Bonferroni correction (pBonferroni < 0.05). (E, F) Enrichment analyses of TWAS for organic annotations within the NHGRI-EBI GWAS Catalog. (E) Enrichment for organic annotations of genes whose gr-expression predicted regional volumes (pFDR < 0.05). Every level represents a organic annotation related to a minimum of 1 gene. The horizontal axis exhibits the p-values (–log10 pFDR) of particular person annotations. Supply information could be present in S3 Table. (F) Relationship between p-values and brain-relatedness of organic annotations. The horizontal axis exhibits bins of p-values (–log10 pFDR). The vertical axis exhibits the fraction of brain-related annotations inside every bin. The p-value on the correlation coefficient was computed by permuting the annotations (estimated from 10,000 random samples). (G, H) Heatmaps of inter-regional TWAS between gr-expression and regional volumes. (G) Absolute numbers of associations. Numbers of genes whose gr-expression in 1 area (columns) predicted (pFDR < 0.05) the amount of one other area (rows). Supply information could be present in S4 Table. (H) Overlap coefficients. Variety of genes that had been widespread to each intra-regional and inter-regional associations in G, normalized by the scale of the smaller of the intra- and inter-regional gene units. FDR, false discovery charge; GWAS, genome-wide affiliation research; SNP, single-nucleotide polymorphism; TWAS, transcriptome-wide affiliation research.
Our first TWAS inferred associations between gray-matter volumes and gr-expression of the identical areas. To attenuate the confounders of genetic ancestry, we restricted our analyses to the “White British” pattern of the UK-Biobank cohort (S1 Table) [37]. We due to this fact carried out TWAS on 39,565 individuals (52.2% feminine, 64.3 ± 7.7 years outdated), with covariates of genetic ancestry, intercourse, and age (Strategies).
We recognized 1,065 associations (of 778 distinctive genes) between gr-expression and the volumes of 8 mind areas (pFDR < 0.05, Fig 3B and S1 and S2 Tables). The variety of regional associations various from 68 genes within the amygdala to 205 genes within the cerebellar hemisphere. Many genes that had been discovered on this evaluation, together with CRHR1, ARL17A, NSF, and OGFOD2, have been implicated in earlier GWAS of regional mind volumes, and have additionally been linked to mind problems, together with epilepsy, schizophrenia, and mind most cancers [62–64].
TWAS reinforce GWAS associations and uncover novel associations
To straight present the methodological benefits of gene-based TWAS, we straight in contrast these research to SNP-based GWAS. We made this comparability in 3 complementary methods.
Direct relationship to GWAS. First, we carried out a GWAS on the identical pattern and in contrast our TWAS associations for particular person genes to GWAS outcomes for the SNPs that fashioned a part of corresponding fashions of gr-expression. These comparisons had been dominated by many-to-many relationships between genes and SNPs, as a result of a number of SNPs sometimes affiliate with the gr-expression of a single gene, and equally, a single SNP can contribute to the gr-expression of a number of genes. The correlations between GWAS and TWAS p-values had been reasonable however statistically vital (0.275 ≤ rspearman ≤ 0.373, p < 0.001 for all areas, Fig 3C left, S1 Fig). To deal with the strongest TWAS and GWAS indicators, we filtered these information in a means that retained the bottom p-value SNP for every gene and, concurrently, the bottom p-value gene for every SNP. This course of resulted in a lot stronger and strictly one-to-one relationships (0.479 ≤ rspearman ≤ 0.583, p < 0.001 for all areas, Fig 3C proper and S1 Fig). Collectively, these outcomes present that gene-based TWAS associations are associated to, but additionally distinct from, SNP-based GWAS associations.
Statistical energy. Second, we investigated the character of those variations by contrasting the variety of associations detected by TWAS and GWAS. The excessive multiple-testing burden of GWAS sometimes requires strict genome-wide Bonferroni corrections. In contrast, the comparatively smaller variety of statistical checks in TWAS leads to a decrease a number of testing burden, and the anticipated polygenic associations of many phenotypes make it widespread to undertake much less strict false discovery charge (FDR) corrections as an alternative choice to Bonferroni [48]. In our analyses, TWAS beneath each corrections recognized many extra genes than the corresponding GWAS (Fig 3D). Particularly, beneath FDR correction, TWAS detected associations of 673 distinctive genes (pFDR < 0.05) that lacked GWAS associations of corresponding SNPs (pBonferroni < 0.05). Many of those genes have been beforehand linked to brain-related problems, together with Alzheimer’s illness (WDR12, AGFG2, and CDK5RAP3), schizophrenia (SRA1, WDR55, CORO7, DDAH2, PCDHA8), autism spectrum dysfunction (MAPK3, PCDHA13), and main depressive dysfunction (ZMAT2 and ITIH4) [65–74]. Individually, beneath Bonferroni correction, TWAS detected associations of 110 distinctive genes (pBonferroni < 0.05) that lacked GWAS associations of corresponding SNPs (pBonferroni < 0.05). These outcomes present that TWAS discovers associations of many genes which might be undetected with GWAS.
Neurobiological interpretability. Third, to interpret the operate of found genes extra systematically, we examined the enrichment of our TWAS outcomes utilizing the NHGRI-EBI GWAS Catalog, a catalog of gene annotations curated from all human GWAS within the present literature [75]. We found 276 enriched organic annotations at pFDR < 0.05 (Fig 3E and S3 Table) and located that brain-related annotations had been more likely to be enriched than different annotations within the catalog (p < 0.001). Furthermore, along with the general enrichment for brain-related annotations, we discovered a powerful optimistic correlation between the p-values of the enrichment and the fraction of found brain-related annotations (rspearman = 0.964, p < 0.001, Fig 3F). In different phrases, we discovered that essentially the most enriched gene annotations had been primarily mind associated. S2 Fig exhibits that these enrichments had been replicable with a Bonferroni correction on TWAS associations. Collectively, these outcomes present the neurobiological relevance of our discoveries.
TWAS uncover associations of genetically regulated gene expression in a single mind area with gray-matter volumes of different areas
Individually, we constructed on our region-specific TWAS findings to check for associations between gr-expression in a single mind area and gray-matter volumes of different areas. Such associations are undefined for SNPs (as a result of all cells share the identical genome), however are interpretable for gr-expression (due to recognized inter-regional similarities in gene expression and group [15,20,23,25]). In sensible phrases, these analyses additionally assist to find associations of regional volumes with genes for which these areas at the moment lack fashions of gr-expression (Fig 1B).
Inter-regional TWAS found between 73 and 209 (median 133) associations (pFDR < 0.05) of gr-expression in a single area with the amount of one other area (Fig 3G and S4 Table). gr-Expression within the amygdala and anterior cingulate had the biggest variety of such associations (Fig 3G, columns) relative to the entire obtainable variety of gr-expression fashions in every area (Fig 1B). For instance, the gr-expression of FOXO3 within the anterior cingulate predicted the volumes of all 8 areas. This gene has been strongly linked to wholesome getting older in numerous human populations [76–78]. In contrast, the amount of putamen was predicted by the biggest variety of genes from different areas (Fig 3G, rows). A number of of those genes—together with MYLK2, KTN1, DCC, BCL2L1, TPX2, and HELZ—had been related to putamen quantity in earlier research [16,79–84]. Specifically, in our research, the gr-expression of MYLK2 and KTN1 predicted putamen quantity in all areas that had gr-expression fashions of those genes (in 8 and 4 areas, respectively). In different instances, gr-expression of some genes in lots of areas predicted volumes of many different areas. For instance, the gr-expression of LRRC37A2 in all 8 areas predicted volumes of all areas besides putamen and caudate. Equally, gr-expression of MAPT within the cerebellar hemisphere predicted all volumes besides putamen and caudate. Each LRRC37A2 and MAPT have been linked to Parkinson’s illness, and MAPT encodes for tau and has been effectively studied within the Alzheimer’s illness literature [50,85,86].
We lastly quantified the overlap between intra-regional and inter-regional associations. A heatmap of overlap coefficients of those associations fashioned 3 anatomically distinct groupings of cortical, basal ganglionic, and limbic areas (Fig 3H). These groupings present that the volumes of anatomically comparable areas usually tend to share gene associations or, alternatively, that genes from one area are related to volumes of anatomically comparable areas. S2 Fig exhibits that these groupings had been replicable with a Bonferroni correction on TWAS associations.
Collectively, these outcomes recommend a powerful relationship between gr-expression profiles of anatomically comparable mind areas and, extra usually, present the utility of inter-regional TWAS of neuroimaging phenotypes.
Genetically regulated gene expression hyperlinks regional volumes with medical phenotypes
We subsequent moved past literature-based annotations to check whether or not gr-expression associations can hyperlink regional volumes with medical phenotypes. To attain this, we built-in our outcomes with a separate TWAS on a pattern of 70,439 individuals in BioVU, a biobank that incorporates DNA samples and de-identified digital well being data for sufferers at Vanderbilt College Medical Heart [53,87,88]. Scientific phenotypes derived from digital well being data in BioVU had been represented by phenotype codes extracted from Worldwide Classification of Illnesses (ICD-9) billing codes. The BioVU TWAS used the identical Joint-Tissue Imputation fashions to estimate gr-expression and to find medical associations (Fig 4A). In what follows, we filtered this medical TWAS to deal with 156 brain-related medical phenotypes. We then in contrast associations of regional gr-expression with these phenotypes to associations in our inter-regional neuroimaging TWAS.
Fig 4. Affiliation of gr-expression with each neuroimaging and medical phenotypes.
(A) Pipeline for BioVU TWAS: transcriptome-wide affiliation research of regional gr-expression and medical phenotypes from the BioVU Biobank. Prime left: Inputs to TWAS comprise digital well being data and DNA samples of the identical individuals. Prime heart: Scientific phenotypes are extracted from ICD-9 codes current in digital well being data. Backside left and heart: Regional gr-expression is estimated from DNA samples of the identical individuals. Proper: Scientific phenotypes and regional gr-expression are mixed within the BioVU TWAS. (B) Heatmap displaying the variety of occasions by which genes (rows) with regional gr-expression (columns) had been linked to each regional volumes and medical phenotypes. Every depend denotes a regional gr-expression that was related (pFDR < 0.05) with each a regional quantity within the UK Biobank TWAS and with a brain-related medical phenotype within the BioVU TWAS. (C) Heatmap displaying the variety of genes with regional gr-expression that linked regional volumes (columns) with medical phenotypes (rows). Every depend denotes a regional gr-expression that was related (pFDR < 0.05) with each a regional quantity within the UK Biobank TWAS and with a brain-related medical phenotype within the BioVU TWAS. (D) Enrichment of medical phenotypes for genes whose gr-expression predicted (pFDR < 0.05) regional volumes (rows) within the UK Biobank TWAS. Every level represents a brain-related medical phenotype related to a minimum of 1 gene. The horizontal axis exhibits the p-values (–log10 pFDR) of particular person phenotypes. Supply information could be present in S5 Table. FDR, false discovery charge; TWAS, transcriptome-wide affiliation research.
We recognized 98 genes whose gr-expression in a selected area related (pFDR < 0.05) with each volumes within the UK Biobank TWAS and with brain-related medical phenotypes within the BioVU TWAS (Fig 4B). There have been 22 genes on this set whose gr-expression in 4 or extra areas linked volumes and medical phenotypes. In earlier GWAS and medical research, these genes have been related to neurogenesis (WNT3) [89,90], neurodevelopmental delays (QRICH1) [91,92], dependancy (HCG27) [93], despair (CCDC71, CYP21A2) [94,95], and different brain-related problems [96,97].
BioVU medical phenotypes that shared associations of gr-expression with regional volumes included quite a lot of nervous system signs and problems together with, most prominently, demyelinating ailments, motor-related signs, and dementia (Fig 4C). A number of HLA genes that play a serious position within the immune response (together with HLA-B/C, HLA-DRB1, and HLA-DRB5) had been related to 2 or extra regional volumes and concurrently with demyelinating ailments, together with a number of sclerosis, a distinguished immune-mediated dysfunction [98]. As well as, genes within the HLA-DR and HLA-DQ households had been related to volumes of the cerebellar hemisphere and hippocampus within the UK Biobank and concurrently with the irregular motion phenotype within the BioVU TWAS. These associations signify candidate causal mechanisms for linking these genes with Parkinson’s illness and different motion problems [99–102]. Genes C4B, MST1, and LRRC37A confirmed comparable patterns of associations, on this means supporting and increasing earlier hyperlinks to motor problems [86,103–105].
Individually, we recognized 9 brain-related medical phenotypes that had been enriched (pFDR < 0.05) for genes whose gr-expression predicted regional volumes (Fig 4D and S5 Table). Most of those phenotypes had been enriched for genes that predicted a number of regional volumes. For instance, myoclonus was enriched for genes that predicted volumes of 6 areas, whereas a number of sclerosis and lack of coordination had been enriched for genes that predicted volumes of 4 areas. Additional, senile dementia was enriched for genes that predicted hippocampal and cerebellar volumes, whereas speech disturbances was enriched for genes that predicted anterior cingulate quantity. The vast majority of motor-related medical phenotypes had been enriched for genes that predicted volumes of the cerebellum, a well known heart of motor management. S3 Fig exhibits that our affiliation and enrichment analyses had been replicable with a Bonferroni correction on TWAS associations.
Total, these outcomes present that associations of gr-expression with phenotypes at totally different organic scales could be mixed to disclose genes that hyperlink regional volumes and medical phenotypes. Regardless of variations in samples and phenotype modalities, we recognized a big overlap within the 2 TWAS between associations with regional gr-expression. Moreover, we discovered proof in associated literature that helps associations between regional volumes and an array of brain-related problems. Collectively, these findings spotlight the built-in relationships between gene expression and mind phenotypes and the implications of those relationships for the research of brain-related problems.
Polygenic fashions of genetically regulated gene expression detect associations in a small neuroimaging information set
Latest research have proven the potential of mixing the gr-expression of a number of genes into polygenic fashions to enhance the prediction of phenotypes (Fig 5A) [106–108]. Such polygenic fashions could also be significantly related for extremely polygenic phenotypes of mind anatomy and exercise. They will additionally seize the polygenic nature of structural and practical MRI phenotypes and additional cut back the variety of statistical checks.
Fig 5. Associations of polygenic gr-expression with neuroimaging phenotypes.
(A) A framework for polygenic modeling of regional phenotypes. Polygenic gr-expression was outlined because the imply normalized gr-expression of genes that had been nominally related to phenotypes at p < 0.001, uncorrected. To make the imply effectively outlined, the indicators of gr-expressions with detrimental associations had been reversed. Mannequin efficiency was evaluated utilizing permutation testing. (B) Consultant scatter plots of neuroimaging phenotypes and polygenic gr-expression. Factors signify people, and colours denote areas as labeled in C. (C) Pearson correlation coefficients between neuroimaging phenotypes and polygenic gr-expression (sq. factors), polygenic gr-expression from permutation checks (field plots; n = 10,000), and greatest single-gene gr-expression from TWAS (spherical factors). Stars signify p-values of polygenic associations with permutation testing (* p < 0.05, ** p < 0.005, *** p < 0.0005). Supply information could be present in S1 Data. (D) Comparability of p-values (−log10 pFDR) from polygenic gr-expression associations and greatest single-gene TWAS. Colours denote areas, whereas strains denote p = 0.05. DLPFC, dorsolateral prefrontal cortex; FDR, false discovery charge; TWAS, transcriptome-wide affiliation research.
Right here, we examined the ability of such analyses utilizing the HCP [54], a small however distinguished neuroimaging genomic information set with high-quality practical MRI information. To attenuate the confounders of genetic ancestry, we restricted our evaluation of this information set to a pattern of 657 non-twins of European genetic ancestry. Our analyses thought of regional quantity phenotypes, in addition to a consultant set of practical MRI phenotypes. The practical MRI phenotypes monitor properties of regional exercise (amplitude of low-frequency fluctuations [109]), within-regional correlation (regional homogeneity [110]), and common inter-regional correlated exercise (imply coactivity [111]). Particularly, amplitude displays the ability of low-frequency exercise, homogeneity displays the extent of intra-regional correlated exercise, whereas coactivity complementarily displays the extent of inter-regional correlated exercise (Strategies). These phenotypes present insights into the group of mind exercise and have been extensively studied in neuroimaging genomics [26,27,112–114].
We first carried out a single-gene TWAS on these phenotypes. The comparatively small measurement of our pattern, nevertheless, essentially resulted in few associations that survived corrections for a number of comparisons. For instance, and in distinction to the UK Biobank TWAS, most regional phenotypes on this evaluation confirmed no associations at pFDR < 0.05. Furthermore, as anticipated, the strongest associations on this pattern had a lot larger p-values (greatest pFDR = 0.017) than the strongest associations within the UK Biobank TWAS (greatest pFDR = 1.34 × 10−21).
We then estimated polygenic gr-expression because the imply normalized gr-expression of genes that had nominal TWAS associations with phenotypes (p < 0.001, uncorrected). We examined associations of polygenic gr-expression in opposition to null associations of equivalently estimated polygenic gr-expression on information with randomized (permuted) task of phenotypes to topics.
Associations of polygenic gr-expression with phenotypes had imply ± commonplace deviation r = 0.434 ± 0.113 (Figs 5B and S4). For regional quantity and homogeneity phenotypes, these associations tended to be larger than null associations (p < 0.05) and have decrease p-values than single-gene associations (Fig 5C and 5D and S1 Data). In contrast, for amplitude and coactivity phenotypes, these associations didn’t are typically larger than null associations and had comparable p-values as single-gene associations (Fig 5C and 5D and S1 Data). Observe additionally that polygenic gr-expression estimated from extra chosen genes tended to have larger associations in absolute phrases and relative to the null associations (S5 Fig). Collectively, these analyses present that polygenic modeling can additional enhance the power of TWAS to deduce associations of teams of genes with advanced phenotypes.
Replicability of estimated genetically regulated gene expression and TWAS
We lastly examined the replicability of our analyses in 3 complementary methods.
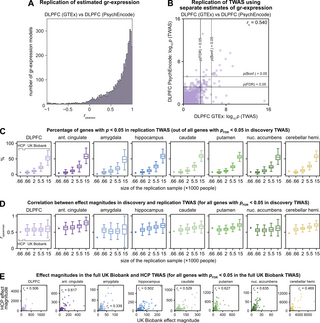
First, we examined the replicability of gr-expression fashions by evaluating the estimated gr-expression of the DLPFC utilizing fashions skilled on 2 distinct postmortem samples: our foremost pattern from GTEx and an impartial replication pattern from PsychEncode [43]. We discovered that fashions skilled on the two samples had extremely comparable patterns of gr-expression (rpearson of gr-expression: median 0.799, Q1–Q3 0.559–0.917, Fig 6A). Likewise, we discovered comparable TWAS of those fashions with DLPFC volumes (rspearman = 0.540, p < 0.001, Figs 6B and S6). These outcomes recommend that our framework for estimating gr-expression is strong to the coaching information, a minimum of for sufficiently giant samples.
Second, we examined the replicability of affiliation p-values and magnitudes within the UK Biobank utilizing the impartial HCP TWAS. As we noticed above, the small HCP pattern produced virtually no associations at pFDR < 0.05. Correspondingly, we discovered {that a} small share of associations with pFDR < 0.05 within the UK Biobank had been additionally current on the nominal threshold of p < 0.05 within the HCP TWAS (median 7.00%, Q1–Q3 4.53%–7.75%, Fig 6C). In contrast, the magnitudes of particular person associations are strongly correlated with p-values (rspearman between magnitudes and −log10p: median 0.783, Q1–Q3 0.773–0.786, S6 Fig) however, not like p-values, are comparatively impartial of the pattern measurement [115]. Correspondingly, we discovered persistently sturdy correlations between magnitudes of associations that handed pFDR < 0.05 within the UK Biobank TWAS (rspearman: median 0.518, Q1–Q3 0.486–0.622, all p < 0.005, Figs 6D, 6E and S6).
Fig 6. Replicability of estimated genetically regulated gene expression and TWAS.
(A, B) Replication of estimated gr-expression skilled on impartial PsychEncode information. (A) Histogram of correlations between gr-expression of the DLPFC estimated with fashions skilled on GTEx information and impartial PsychEncode information. (B) Scatter plot of TWAS associations based mostly on gr-expression of the DLPFC estimated with fashions skilled on GTEx information and impartial PsychEncode information. Every level denotes p-values of associations between estimated gr-expression and DLPFC gray-matter volumes within the white-British pattern of the UK-Biobank cohort. (C–E) Replication of genes that handed pFDR = 0.05 in discovery TWAS of gray-matter volumes. (C) Percentages of genes that had been replicated at nominal p < 0.05 in replication TWAS. Supply information could be present in S2 Data. (D) Correlations between impact magnitudes of genes within the replication and discovery TWAS. Dots denote analyses on the complete UK Biobank (discovery) and HCP (replication) samples. Field plots denote analyses of discovery-replication splits of the white-British UK-Biobank pattern, ordered from small to giant replication samples. Every field plot was estimated from 300 random splits. (E) Scatter plots of impact magnitudes within the UK Biobank and HCP TWAS. Every level denotes impact magnitudes for a gene that confirmed pFDR < 0.05 within the UK Biobank TWAS. DLPFC, dorsolateral prefrontal cortex; FDR, false discovery charge; GTEx, Genotype-Tissue Expression Challenge; HCP, Human Connectome Challenge; TWAS, transcriptome-wide affiliation research.
Third, we repeated these analyses on TWAS of discovery and replication subsets generated from 1,200 random splits of the white-British UK-Biobank pattern (S2 Data). These further analyses confirmed that replication samples of the identical measurement as our HCP pattern (657 individuals) had equally small percentages of replicable associations (median 6.60%, Q1–Q3 6.20%−6.82%) and that bigger samples confirmed a lot larger percentages (Fig 6C). Likewise, these analyses confirmed that replication samples of the identical measurement as our HCP pattern had sturdy correlations between magnitudes of results (rspearman: median 0.575, Q1–Q3 0.568–0.579; all p < 0.001) and that bigger samples confirmed modestly elevated correlations between magnitudes (Figs 6D and S6).
Collectively, these analyses recommend that the estimated gr-expression and magnitudes of TWAS associations had been usually replicable, whereas the p-values of TWAS associations had been replicable in giant replication samples.
Interactive software to facilitate adoption of TWAS in neuroimaging genomics
To extend the accessibility of our outcomes, we created a browser-based software to discover our SNP-based and gene-based associations (https://github.com/nhunghoang/twas-webapp). The applying permits customers to match neuroimaging GWAS with neuroimaging TWAS and with medical TWAS and, on this means, hyperlinks analyses of SNPs, genes, neuroimaging phenotypes, and medical phenotypes. It additionally permits customers to interactively discover associations and offers extra direct gene-based interpretations of SNP-based outcomes.
Dialogue
Abstract
We adopted state-of-the-art strategies from computational genomics to estimate genetically regulated gene expression, or gr-expression, throughout 10 cortical and subcortical mind areas in additional than 40,000 individuals. First, we confirmed that estimates of gr-expression throughout individuals and mind areas recapitulate the neurobiological group of straight assayed gene expression. Second, we confirmed that TWAS based mostly on estimated gr-expression aligned with, and prolonged, associations from corresponding GWAS. Third, we built-in these outcomes with a set of impartial associations between regional gr-expression and brain-related medical phenotypes extracted from digital well being data. Fourth, we confirmed that polygenic fashions of gr-expression can additional enhance the statistical energy of our strategy. Lastly, we confirmed that estimated gr-expression ranges and the magnitudes of TWAS associations had been usually replicable whereas the p-values of TWAS associations had been replicable in giant samples.
Advances
Our research exhibits that gene-based affiliation analyses can bridge gaps in present neuroimaging genomic research. Particularly, the strategy begins to fill the mechanistic hole in conventional GWAS by extending these research to determine associations of phenotypes with regionally particular gr-expression, quite than with regionally agnostic SNPs. Furthermore, the strategy reduces the multiple-testing burden of GWAS by orders of magnitude. Individually, the strategy enhances regional transcriptomic research by extending these research to 1000’s of individuals with obtainable genomes.
We demonstrated the distinctive mixture of those benefits with 3 complementary analyses. First, we confirmed that, like GWAS, our methodology separates individuals by genetic ancestry (Fig 2A). Second, we confirmed that, like regional transcriptomic analyses, our methodology separates mind areas by patterns of gr-expression (Fig 2B). Third, we confirmed sturdy similarities between the inter-regional correlation of gr-expression from the UK Biobank and straight assayed expression from the Allen Human Mind Atlas and the GTEx Challenge (Fig 2C–2K). On this means, we confirmed that our strategy produces neurobiologically interpretable estimates of regionally particular gr-expression in giant human populations.
Our methodology allowed us to straight benchmark gene-based TWAS in opposition to SNP-based GWAS. First, we confirmed that TWAS associations are broadly associated to, but additionally distinct from, GWAS associations (Fig 3C). Second, we confirmed that TWAS can uncover associations of many genes which might be in any other case undetected with GWAS (Fig 3D) and that these discoveries strongly favor brain-related annotations (Fig 3E). Third, we confirmed that inter-regional associations are interpretable and additional enhance the utility of TWAS (Fig 3F). Collectively, these outcomes straight exhibit the conceptual and sensible strengths of TWAS.
Individually, our research constructed on these leads to 3 further methods. First, it built-in estimates of regionally particular gr-expression with gray-matter volumes and brain-related medical phenotypes (Fig 4). Second, it prolonged the single-gene TWAS to construct polygenic fashions of neuroimaging phenotypes (Fig 5). Third, it confirmed that the magnitudes of TWAS associations are usually replicable however that the p-values of TWAS associations are extremely delicate to pattern sizes (Fig 6). These outcomes define a path in the direction of the replicable integration of polygenic gr-expression with advanced neuroimaging and medical phenotypes.
Individually, the research of gr-expression offers distinctive benefits over the research of straight assayed expression as a result of it permits to deal with the steady, genetically regulated features of gene expression with out the necessity to management for the potential confounders of environmental elements and acquisition biases, together with batch results [116]. Equally, an vital benefit of this research relative to group-averaged transcriptomic research is the dearth of evident bias attributable to distance results [60]. This lack of bias arises as a result of the associations are computed over individuals, quite than over mind areas. For instance, whereas we discovered that the noticed inter-regional correlation between estimated gr-expression and straight assayed expression couldn’t be defined solely by distance dependence, a distance-based rationalization wouldn’t invalidate our outcomes as a result of it could replicate organic, quite than artifactual, results.
Limitations
Our strategy has many advantages, but it surely additionally has limitations. First, our analyses nonetheless require giant samples to allow replicable associations (Fig 6C–6E). Nonetheless, the decrease multiple-testing burden of TWAS makes this downside much less acute than for checks of tens of millions of SNPs in GWAS. Equally, very similar to correlations of adjoining SNPs in GWAS, correlations of gr-expression in TWAS, whereas usually smaller and fewer widespread, could make it tough to fine-map causal genes. Future research may undertake mendelian randomization to allow causal inference, though this strategy comes with its personal limitations, together with the issue of accounting for horizontal pleiotropy (the impact of 1 gene on unrelated phenotypes) [117–119]. Lastly, our deal with genes essentially misses the consequences of variants that function by way of means aside from the regulation of gene expression.
Second, relative to spatially particular transcriptomic research, our strategy is restricted to a small variety of mind areas. In future research, we suggest to beat this limitation by modeling recognized relationships between regional and community group [120]. We additionally suggest that the adoption of comparable fashions will enable researchers to combine regional associations with high-resolution single-cell atlases of gene expression and hyperlink these associations to particular cell varieties [121].
Third, our outcomes combine genomic biobanks with obtainable information on genetics, gene expression, neuroimaging, and medical phenotypes. Such integration essentially comes with the challenges of demographic range and matching. Our analyses had been based on European populations and should not essentially generalize to different populations. As genomic, transcriptomic, neuroimaging, and medical information proceed to extend in measurement and scope, it is going to be vital to increase these outcomes to analyses of different populations.
Strategies
Joint-Tissue Imputation fashions
We used Joint-Tissue Imputation fashions of gr-expression that had been beforehand skilled on postmortem gene expression information from the GTEx. On this part, we describe the principle features of high quality management and coaching of those fashions. We refer the readers to the unique research of Joint Tissue Imputation [43] and the GTEx v8 information set [56] for a extra detailed dialogue of those approaches.
Joint-Tissue Imputation fashions estimate gr-expression because the linear mixture of SNPs which might be near the gene of curiosity alongside the linear genome. The coaching of those fashions, due to this fact, required information on tissue-specific gene expression and whole-genome sequencing from the identical individuals. The GTEx v8 information set included these information for mind areas of 838 donors that handed inner GTEx biospecimen quality control [122]. The donors had the next demographics: Age, 21 to 70 years (imply 53); intercourse, 34% feminine; ancestry, 85.3% European American/12.3% African American/1.4% Asian American. The info set contained RNA-seq from ten mind areas. Table 1 summarizes the names of those areas and the variety of samples used to coach fashions in every area. For completeness, it additionally summarizes our definitions of the corresponding areas in volumetric Allen Human Mind Atlas information, and in surface-based UK-Biobank and Human-Connectome Challenge information.
The next steps had been taken to maximise the predictive accuracy of estimation and to reduce confounders. First, the assayed gene expression ranges had been managed for intercourse, sequencing platform, the highest 5 principal elements, in addition to probabilistic estimation of expression residuals, a Bayesian mannequin of hidden confounders [123]. Second, the fashions had been skilled solely on biallelic SNPs that had a minor allele frequency of a minimum of 0.05 and that had been in Hardy–Weinberg equilibrium (p > 0.05), i.e., solely on SNPs that had each ample and steady variation. Third, to scale back the consequences of linkage disequilibrium, extremely correlated (r2 ≥ 0.9) SNPs had been pruned and the fashions had been skilled solely on SNPs close to the gene of curiosity. The optimum threshold for proximity was decided individually for every gene by cross-validation. Lastly, to moreover management for overfitting, the fashions integrated elastic-net regularization and the coaching was based mostly on 5-fold cross-validation.
As a part of the replication evaluation (S6 Fig), we additionally thought of Joint-Tissue Imputation fashions that had been skilled on 415 samples of sequencing and expression information from the DLPFC within the PsychENCODE challenge [57]. These information had been processed and skilled in the identical means as the unique research of Joint-Tissue Imputation. All pretrained fashions can be found on-line at https://doi.org/10.5281/zenodo.3842289 (GTEx-trained fashions) and https://doi.org/10.5281/zenodo.3859065 (PsychEncode-trained fashions).
Genotype-Tissue Expression Challenge (GTEx) and Allen Human Mind Atlas Information
Our analyses of inter-regional correlations (Fig 2) in contrast the estimated gr-expression information described within the earlier part to straight assayed expression information from GTEx and the Allen Human Mind Atlas. This part describes our preprocessing of those latter information units.
We downloaded the newest launch (v8) of the GTEx gene-expression information from https://gtexportal.org/home/downloads/adult-gtex. These information had been acquired from 340 donors (a mean of 199 donors per area). Gene expression ranges had been quantified and normalized by GTEx, and genes had been chosen based mostly on expression thresholds as beforehand described [56].
We downloaded the Allen Human Mind Atlas microarray gene-expression information from https://human.brain-map.org/static/download. The info had been acquired from 6 donors (42 ± 12 years outdated, 1 feminine). Mind-wide gene expression ranges had been quantified and normalized by the Allen Institute, as beforehand described [130].
Our preprocessing of those information adopted present greatest practices [131]. All imputed and straight assayed expression information had been normalized to have zero imply and unit variance throughout areas. As well as, information from the Allen Human Mind Atlas had been filtered to exclude genes whose expression degree didn’t exceed the background sign (as specified by the file PACall.csv). These information had been additionally nonlinearly registered to reference coordinate area [132], assigned to areas with a 2 mm distance threshold, and averaged throughout all obtainable probes and the left and proper hemispheres.
UK Biobank genomic and neuroimaging information
We analyzed information from 45,549 individuals, or all obtainable individuals from the UK Biobank with genome-wide genotyping and neuroimaging volumes. Our pattern had the next demographics: Age, 64 ± 7.7 years outdated; intercourse, 52% feminine; self-reported ethnicity, 96.7% white/0.6% black/1.1% South Asian/0.3% Chinese language/0.5% Combined/0.8% Different (S1 Table). On this part, we describe the principle features of high quality management and processing of those information by the UK Biobank. We refer the readers to the unique publications [17,37,133] for a extra detailed dialogue of those and different questions.
Genome-wide genotype imputation was carried out utilizing information from the Haplotype Reference Consortium [134] as the principle imputation reference panel, in addition to merged UK10K and 1000 Genomes Section 3 information units because the secondary imputation reference panel [135]. The info handed an automatic quality-control pipeline [37]. This pipeline comprised marker-based and sample-based high quality management. Marker-based management included checks for batch results, plate results, deviation from Hardy–Weinberg equilibrium, intercourse results, array results, and sequencing replicability. Individually, sample-based management included checks for unusually excessive fractions of heterozygous or lacking loci, in addition to for mismatch between self-reported intercourse and the depth of sex-chromosome markers.
Our TWAS (Figs 3 and 4) sought to reduce the confounders of genetic ancestry by specializing in the “White British” pattern of the UK-Biobank cohort (39,565 individuals). We adopted UK Biobank analyses to pick out individuals who self-reported as “White British” and who had comparable genetic ancestry based mostly on UK-Biobank principal element evaluation on 147,604 genotype markers (pruned to minimized linkage disequilibrium) over 407,219 unrelated individuals [37]. In contrast, our analyses of genetic ancestral relationships, regional identities, and inter-regional correlations (Fig 2) centered on the remaining ethnically numerous pattern of the UK-Biobank cohort (5,984 individuals).
All UK Biobank neuroimaging information had been processed by the UK Biobank automated brain-imaging pipeline [133]. The pipeline flagged lacking and distorted information, registered photos to widespread reference area, and computed imaging-derived phenotypes. We used phenotypes computed on MRI scans from the primary imaging go to. Our analyses included solely cortical and subcortical mind areas that had obtainable estimates of gray-matter quantity and gr-expression and that handed UK Biobank high quality management exclusion standards (see the unique reference [133] for an in depth dialogue). Volumes of those areas had been computed by the UK Biobank utilizing FreeSurfer cortical and subcortical segmentations [61] (Table 1) and had been averaged throughout the left and proper hemispheres.
Human Connectome Challenge genomic and neuroimaging information
The complete HCP incorporates 1,142 individuals with brain-wide genotyping sequences and neuroimaging information. This cohort had the next demographics: Age 29 ± 3.7 years outdated, 54% females, 149 pairs of monozygotic twins. On this part, we describe our curation of those information to generate a pattern of 657 individuals. We additionally describe the principle features of high quality management and processing of those information by the HCP, and our estimation of phenotypes from these information. We refer the readers to the unique HCP publications [136–139] for a further in depth dialogue of high quality management and information processing.
Genotyping of all individuals was derived from blood or saliva samples. The genotype information comprised possibilities of single-nucleotide variants, estimated utilizing the Illumina Multi-Ethnic World Array. High quality-control procedures of those information included verification of self-reported widespread ancestry for siblings, in addition to zygosity for twins.
As in our analyses of the UK Biobank, we sought to reduce the confounders of genetic ancestry by focusing our affiliation analyses on a pattern of 657 non-twins of European genetic ancestry. We estimated genetic ancestry utilizing the principal elements of genotyping information from this cohort, computed with EIGENSTRAT [140]. We outlined individuals to be of European ancestry once they self-reported as European and once they had comparable genetic ancestry based mostly on principal-component construction [37]. Lastly, we randomly eliminated a single individual from all pairs of monozygotic twins.
We analyzed structural and resting-state practical MRI phenotypes from the HCP. All information had been processed utilizing the HCP minimal preprocessing pipeline [137] and had been handed by way of a standardized high quality management pipeline [138]. Structural MRI acquisitions had been initially reviewed for picture blurriness, movement, and different artifacts. Volumes of those areas had been then estimated utilizing FreeSurfer cortical and subcortical segmentations [61]. FreeSurfer-based reconstructions had been inspected for apparent errors. Individually, resting-state practical MRI acquisitions had been scored for 9 high quality management measures that centered on the temporal signal-to-noise ratio, picture smoothness, in addition to the extent of absolute and relative head movement. The info had been registered with MSM-All [141] and denoised with ICA-FIX [139].
In our research, we computed 3 practical MRI phenotypes on these information. First, we computed the amplitude of low-frequency fluctuations as the entire energy of spontaneous intra-regional exercise throughout the 0.01 to 0.08 Hz vary. Second, we computed regional homogeneity because the imply Pearson correlation between all pairs of intra-regional voxels. Third, we computed imply coactivity because the imply Pearson correlation between the exercise of the area and all different areas of curiosity.
Particular person variation in homogeneity and coactivity strongly correlated with particular person variation of the worldwide sign, the imply exercise of all mind voxels. This variation can replicate artifact but additionally features of vigilance and non-neuronal physiology [142]. To deal with correlation construction unaffected by such properties, we computed these phenotypes after regressing out the worldwide sign from voxel time collection (for homogeneity) or regional time collection (for coactivity).
We computed every phenotype individually for every scan of every individual after which averaged the phenotypes throughout the 4 obtainable scans and the left and proper hemispheres.
Analyses of genetic ancestry, regional identification, and inter-regional correlation construction
We created principal element embeddings of ancestral and regional gr-expression for the ethnically numerous pattern of the UK-Biobank cohort. We first constructed a 3D array of 5,984 individuals × 1,892 genes × 10 areas, the place individuals comprised the UK-Biobank pattern (S1 Table), and genes comprised all genes with obtainable estimates of gr-expression in all the ten GTEx areas. We then analyzed reshaped variations of this array. First, to extract ancestral construction, we computed principal elements of the 5,984 × 18,920 matrix of brain-wide gr-expression throughout individuals. Second, to extract regional construction, we computed principal elements of the 59,840 × 1,892 matrix of region-specific gr-expression throughout individuals.
We likewise in contrast inter-regional correlations of expression on subsets of genes that concurrently had obtainable expression within the UK-Biobank, GTEx, and Allen Human Mind Atlas information. These subsets ranged from 2,837 to 4,220 genes (median 3,642) and differed for every area of curiosity as a result of every area had a definite set of obtainable gr-expression fashions. For all pairs of areas, we computed inter-regional coexpression utilizing the subsets of genes that had expression information in each areas. Lastly, we averaged the inter-regional coexpression matrices throughout all individuals in every information set.
To check for distance results, we computed Spearman correlation coefficients between inter-regional coexpression and Euclidean distance between centroids of areas within the volume-based parcellation (Table 1). To check the consequences of distance on the similarity of inter-regional coexpression, we generated 10,000 coexpression matrices with permuted ranks and empirical Spearman correlations with Euclidean distance.
Affiliation of genetically regulated gene expression with neuroimaging phenotypes
Transcriptome-wide affiliation research (TWAS).
We estimated associations of gr-expression and neuroimaging phenotypes utilizing unusual least sq. regression fashions, with covariates of genetic ancestry, intercourse, and age. We adopted widespread practices for addressing inhabitants stratification by modeling genetic ancestry by the highest 40 principal elements of the genotypes in every pattern. We used the precomputed principal elements for the UK Biobank [37] and EIGENSTRAT [140] to compute principal elements for the HCP. We examined associations between the gr-expression and quantity of the identical area (intra-regional TWAS, S2 Table) and between the gr-expression of 1 area and the volumes of different areas (inter-regional TWAS, S4 Table).
Genome-wide affiliation research (GWAS).
We used REGENIE [143] to carry out GWAS on the regional gray-matter volumes for the white-British pattern of the UK-Biobank cohort. REGENIE is a machine-learning methodology for becoming genome-wide regressions to advanced phenotypes, significantly for big samples with a number of phenotypes of curiosity. We first filtered (straight genotyped and imputed) autosomal SNPs with a minor allele frequency higher than 1% and an info rating higher than 0.2 based mostly on the complete UK-Biobank cohort of roughly 500,000 individuals (info rating denotes the fraction of information at an imputed marker that roughly equates to completely noticed genotype calls [37]). We then set imputed SNPs utilizing a hard-call threshold of 0.01 and, with respect to our pattern of curiosity, filtered the SNPs with a minor allele frequency higher than 1%, missingness lower than 5%, and Hardy–Weinberg equilibrium take a look at p < 10−5. We carried out GWAS on the remaining 8,072,589 SNPs, after regressing out age, intercourse, and the highest 40 principal elements.
Comparability of genome-wide and transcriptome-wide associations.
Joint-Tissue Imputation fashions of gene gr-expression can, normally, comprise a number of SNPs. Equally, one SNP can, normally, be a part of a number of gr-expression fashions. We used this data to map GWAS-derived SNP associations to TWAS-derived gene associations (Fig 3C). We made this mapping utilizing 2 approaches:
Many-to-many mapping: On this mapping, we linked every TWAS-derived gene affiliation with all SNPs that comprised the gr-expression mannequin of that gene. Equivalently, we linked every GWAS-derived SNP affiliation to all of the gr-expression fashions of which that SNP was a component.
One-to-one mapping: We subsequent filtered the many-to-many mapping in 2 steps. First, we filtered TWAS-derived gene associations to protect hyperlinks to the strongest obtainable GWAS-derived SNP affiliation. Second, we filtered the remaining GWAS-derived SNP associations to protect hyperlinks to the strongest remaining TWAS-derived gene affiliation. This two-step filtering of many-to-many associations due to this fact assured one-to-one relationships.
Polygenic modeling affiliation research.
We modeled associations between polygenic gr-expression and phenotypes with covariates of genetic ancestry, intercourse, and age. First, we chosen genes that had nominal TWAS associations with phenotypes (p < 0.001, uncorrected). Second, we averaged the normalized gr-expression of those chosen genes (reversing the signal of gr-expression that had detrimental associations). Third, we computed the Pearson correlation coefficient between these averaged gr-expression and phenotypes. Lastly, we repeated this course of 10,000 occasions on information that had randomized (permuted) task of phenotypes to topics, however the identical values of particular person phenotypes and gr-expression.
Gene-set enrichment analyses
We carried out gene-set enrichment evaluation for organic annotations from the NHGRI-EBI GWAS catalog [75] (Fig 3E). Every phenotype on this catalog is located throughout the Experimental Issue Ontology, a normal ontology that features phrases from a number of extra specialised ontologies and describes a variety of measurements, together with wholesome and diseased phenotypes. All annotations replicate findings from curated GWAS analyses.
We used a semi-automated pipeline to detect brain-related phrases on this ontology in 2 steps. In step one, we flagged every time period as brain-related if phrases in its ontology tree included a minimum of one of many following phrase segments: nerv, neur, cogn, psyc, ment, brai. Second, 2 authors (NH and MR) manually and independently checked these candidate phrases to substantiate or exclude their brain-related nature. S3 Table lists the phenotypes that had been enriched for TWAS genes at pFDR < 0.05 and likewise lists their brain-relatedness indicator.
We carried out gene-set enrichment analyses for medical phenotypes within the BioVU TWAS, a database of associations between genetically regulated gene expression and medical phenotypes derived from the Vanderbilt College Biobank. For these analyses, we thought of 70,439 individuals of European ancestry. Phenotypes had been represented as phenotype codes based mostly on ICD-9 codes [144]. We restricted our analyses to psychological problems and neurological phenotype code classes and included all gene-phenotype pairs that confirmed associations within the BioVU TWAS at pFDR < 0.05.
We carried out gene-set enrichment analyses utilizing WebGestalt [145,146]. In each instances, we used a hypergeometric null mannequin to check the enrichment of genes that had TWAS associations of pFDR < 0.05 in opposition to a reference set of all genes within the TWAS (in different phrases, in opposition to all genes with related gr-expression fashions that handed baseline efficiency thresholds).
Supporting info
S1 Fig. UK Biobank TWAS outcomes for all thought of mind areas. Left.
TWAS of gr-expression and mind volumes for all areas. Every level denotes an affiliation between the person variation of gr-expression of a gene and quantity in the identical area. The horizontal axis denotes the chromosome location of particular person genes. The vertical axis denotes–log10 p-values. Stable-color factors signify associations that handed pFDR = 0.05 or pBonferroni = 0.05 (horizontal strains). Proper. Associations between SNP-based GWAS and gene-based TWAS for all areas. Left: Scatter plots of p-values (–log10 p) for associations of all genes and SNPs. These plots protect all genes and SNPs however lack the one-to-one relationship between genes and SNPs. Proper: Corresponding scatter plots of the best-performing genes and SNPs. Every gene in TWAS matches with its best-performing SNP in GWAS. Equally, every SNP in GWAS matches with its best-performing gene in TWAS. These plots present one-to-one relationships however exclude many genes and SNPs.
https://doi.org/10.1371/journal.pbio.3002782.s001
(TIFF)
S2 Fig. Results of Bonferroni correction on enrichment analyses and inter-regional TWAS.
(A, B) Results of Bonferroni correction on enrichment analyses within the NHGRI-EBI Catalog. (A) Enrichment for organic annotations of genes whose gr-expression predicted regional volumes (pBonferroni < 0.05). Every level represents a organic annotation related to a minimum of 1 gene. The horizontal axis denotes the p-values (–log10 pFDR) of particular person annotations. (B) Comparability of pFDR for organic annotations of genes whose gr-expression predicted regional volumes beneath FDR and Bonferroni corrections. (C, D) Results of Bonferroni correction on inter-regional associations between gr-expression and regional mind volumes. (C) Absolute numbers of associations. Numbers of genes whose gr-expression in a single area (columns) predicted (pBonferroni < 0.05) the amount of one other area (rows). (D) Overlap coefficients. Variety of genes that had been widespread to each intra-regional and inter-regional associations in C, normalized by the scale of the smaller of the intra- and inter-regional gene units.
https://doi.org/10.1371/journal.pbio.3002782.s002
(TIFF)
S3 Fig. Results of Bonferroni correction on associations of gr-expression with each neuroimaging and medical phenotypes.
(A) Heatmap displaying the variety of occasions by which genes (rows) with regional gr-expression (columns) had been linked to each regional volumes and medical phenotypes. Every depend denotes a regional gr-expression that was related to each a regional quantity within the UK Biobank TWAS and with a brain-related medical phenotype within the BioVU TWAS (pBonferroni < 0.05). (B) Heatmap displaying the variety of genes with regional gr-expression that linked regional volumes (columns) with medical phenotypes (rows). Every depend denotes a regional gr-expression that was related to each a regional quantity within the UK Biobank TWAS and with a brain-related medical phenotype within the BioVU TWAS (pBonferroni < 0.05). (C) Enrichment of medical phenotypes for genes whose gr-expression predicted regional volumes (rows) within the UK Biobank TWAS (pBonferroni < 0.05). Every level represents a brain-related medical phenotype related to a minimum of 1 gene. The horizontal axis denotes the p-values (–log10 pFDR) of particular person phenotypes. (D) Comparability of pFDR for medical phenotypes of genes whose gr-expression predicted regional volumes beneath FDR and Bonferroni corrections.
https://doi.org/10.1371/journal.pbio.3002782.s003
(TIFF)
S4 Fig. Associations of polygenic gr-expression with neuroimaging phenotypes.
Scatter plots of polygenic gr-expression and neuroimaging phenotypes. The horizontal axis exhibits values of noticed phenotypes, and the vertical axis denotes values of polygenic gr-expression. Factors signify single people.
https://doi.org/10.1371/journal.pbio.3002782.s004
(TIFF)
S5 Fig. Affiliation of gene numbers with p-values in polygenic fashions.
Scatter plots displaying the variety of genes in every polygenic mannequin and mannequin p-values (–log10 pFDR from permutation testing). Every plot exhibits a definite phenotype. Colours denote mind areas as in Fig 5.
https://doi.org/10.1371/journal.pbio.3002782.s005
(TIFF)
S6 Fig. Replicability of estimated genetically regulated gene expression and TWAS.
(A) Left. Inside-regional associations of gr-expression and gray-matter volumes for the DLPFC, based mostly on gr-expression fashions skilled on GTEx and PsychEncode information. Every level denotes an affiliation between the person variation of gr-expression and quantity in the identical area. The horizontal axis exhibits the chromosome location of particular person genes. The vertical axis exhibits the p-values (–log10 p) of associations. Stable-color factors present associations that handed pFDR = 0.05 or pBonferroni = 0.05 (horizontal strains). (A) Proper. Associations between SNP-based GWAS and gene-based TWAS for the DLPFC, based mostly on gr-expression fashions skilled on GTEx and PsychEncode information. Left: Scatter plots of p-values (–log10 p) for associations of all genes and SNPs. These plots protect all genes and SNPs however lack the one-to-one relationship between genes and SNPs. Proper: Corresponding scatter plots for the best-performing genes and SNPs. Every gene in TWAS matches with its best-performing SNP in GWAS. Equally, every SNP in GWAS matches with its best-performing gene in TWAS. These plots present one-to-one relationships however exclude many genes and SNPs. (B) Scatter plots of impact magnitudes and p-values (–log10 p) for the UK Biobank TWAS of regional gray-matter volumes. Dots denote associations for all genes from the TWAS. Observe the double-logarithmic scale. (C) Correlations between impact magnitudes of all gene associations within the replication and discovery TWAS of regional gray-matter volumes. Dots denote analyses on the complete UK Biobank (discovery) and HCP (replication) samples. Field plots denote analyses of discovery-replication splits of the white-British UK-Biobank pattern, ordered from small to giant replication samples. Every field plot was estimated from 300 random splits of the white-British UK-Biobank pattern. Fig 6D exhibits the same plot, however filtered to incorporate solely genes that handed pFDR < 0.05 within the discovery TWAS.
https://doi.org/10.1371/journal.pbio.3002782.s006
(TIFF)
S1 Data. Affiliation of polygenic gr-expression with neuroimaging phenotypes (Fig 5C).
This information set (hdf5 file) incorporates arrays for reproducing associations in Fig 5C. It particularly incorporates single-gene correlations (file key twas-pearsons), poly-gene correlations (file key poly-pearsons), permutation-test correlations (file key poly-null-pearsons), and order of regional phenotype in these arrays (file key reg-phen-order).
https://doi.org/10.1371/journal.pbio.3002782.s012
(HDF5)
S2 Data. Replication of results and p-values (Fig 6C and 6D).
This information desk incorporates the replication fractions of discovery TWAS genes in Fig 6C (column replication_fraction). It additionally incorporates the correlations between impact magnitudes for these discovery-replication TWAS pairings in Fig 6D (column effect_spearman). TWAS are identifiable by their regional quantity phenotype of curiosity (column area), the replication cohort and measurement (column cohort), and the random-sample iteration (column iteration).
https://doi.org/10.1371/journal.pbio.3002782.s013
(CSV)











{kind=link}